THE IMMUNOLOGY & MICROBIOLOGY DEPARTMENT
In this issue of the newsletter, we will be highlighting the research being carried out by the Immunology & Microbiology Department at DDI, led by Dr. Rasheed Ahmad.
RESEARCH HIGHLIGHT
Discover the latest research being carried out at DDI
Irf3 Is A Key Regulator Of Metabolic Inflammation, Insulin Resistance And Disruption Of Glucose Homeostasis
Published on 01/06/2019
Obesity/overnutrition is associated with a state of chronic low-grade inflammation and its downstream metabolic effects greatly increase the risk of developing metabolic syndrome including Type 2 Diabetes Mellitus (T2DM), cardiovascular disease, fatty liver disease and neuropathy. The role of metabolic inflammation in T2DM and its related diseases is now clear, although the pathways that promote and sustain the inflammation in obesity remain unclear. We must increase our understanding of the factors that are specifically involved in T2DM if we hope to stop metabolic disease while also avoiding complete immunosuppression. Groups led by Dr. Rasheed Ahmad here at DDI, and Professor Evan Rosen at the Harvard Medical School have focused on the role of the transcription factor IRF3 (Interferon regulatory factor 3) in promoting obesity, insulin resistance and inflammation. Together they showed that mice lacking IRF3 in all tissues (i.e., global IRF3 knockout, or KO) have increased brown fat activation, enhanced resistance to obesity, better glucose tolerance, and reduced circulating and local inflammatory markers (Figs.1-2 in Kumari et al, 2016).
In later work initiated by Dr. Ahmad’s group, it was shown that cooperative activation of inflammatory cytokines by fats and TNF-α is mediated by IRF3 (Fig. 3-4: Ahmad et al, 2018). The chemokine CCL2 (also known as MCP-1) is a key regulator of monocyte infiltration into fat tissue, which plays a central role in the disease characteristics of obesity-associated inflammation and insulin resistance. They showed that human monocytic cells and macrophages produced enormous amount of CCL2 in the presence of TNF-α and saturated free fatty acid (FFA) palmitate, a major component of high fat diets. Mechanistically, we have proved that the TNF-α/palmitate mediated overproduction of CCL2 is IRF3-dependent. Diet-induced C57BL/6 obese mice with high FFAs levels showed a strong correlation between TNF-α and CCL2 in plasma and adipose tissue and, as expected, also showed increased adipose tissue macrophage accumulation compared with lean mice. Similar results were observed in the adipose tissue samples from obese humans. Overall, our findings support a model in which elevated FFAs in obesity create an environment for TNF-α to trigger CCL2 production via the TLR4/TRIF/IRF3 signaling cascade, representing a potential contribution of FFAs (high fat diet components) to metabolic inflammation.
Our findings of these two studies provide insight into the major functional features of IRF3 in the development of metabolic inflammation/T2D which may be useful to develop novel therapeutics that interrupt IRF3-associated inflammatory pathways, and thus promote metabolic health.
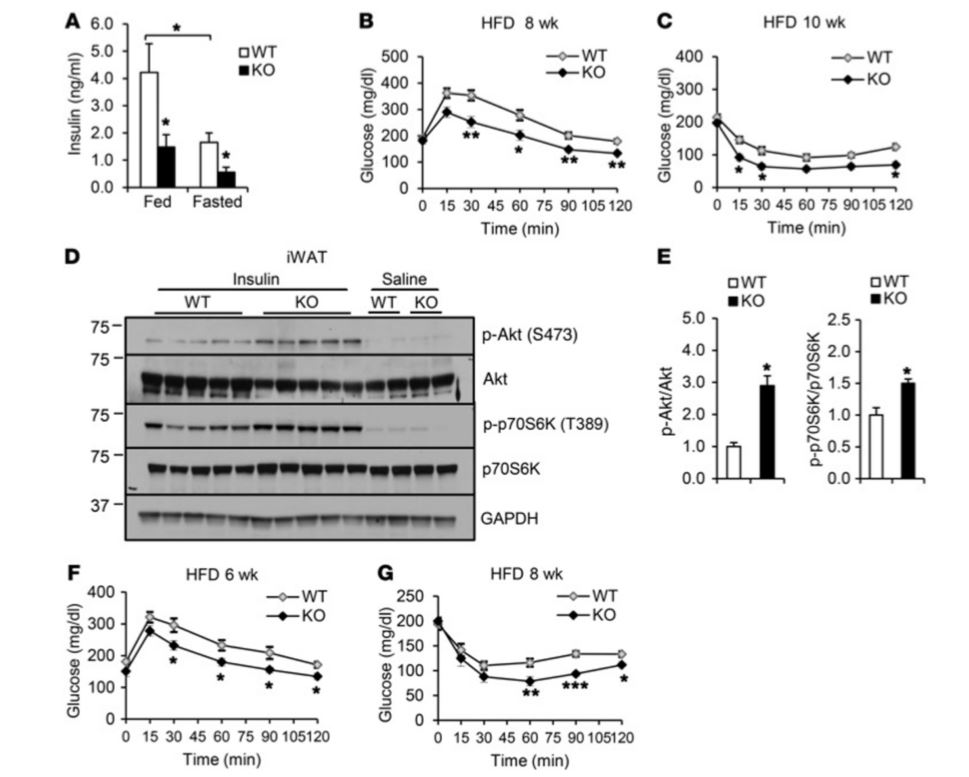
Figure 3 Irf3–/– mice show improved insulin sensitivity on HFD (High Fat Diet). (A) Fed and fasting plasma insulin levels in Wild Type (WT) and Irf3–/– mice on 16 weeks of HFD (n = 7–8 per genotype). (B) Glucose tolerance test after 8 weeks on HFD. (C) Insulin tolerance test after 10 weeks on HFD. (D) Insulin-stimulated Akt and p70S6K phosphorylation in iWAT (inguinal White Adipose Tissue) of WT and Irf3–/– mice after 16 weeks on HFD. (E) Quantification of Western blot in D. (F) Glucose tolerance testing of WT and Irf3–/– mice after 6 weeks of HFD, prior to weight divergence. (G) Insulin tolerance testing of WT and Irf3–/– mice after 8 weeks of HFD, prior to weight divergence
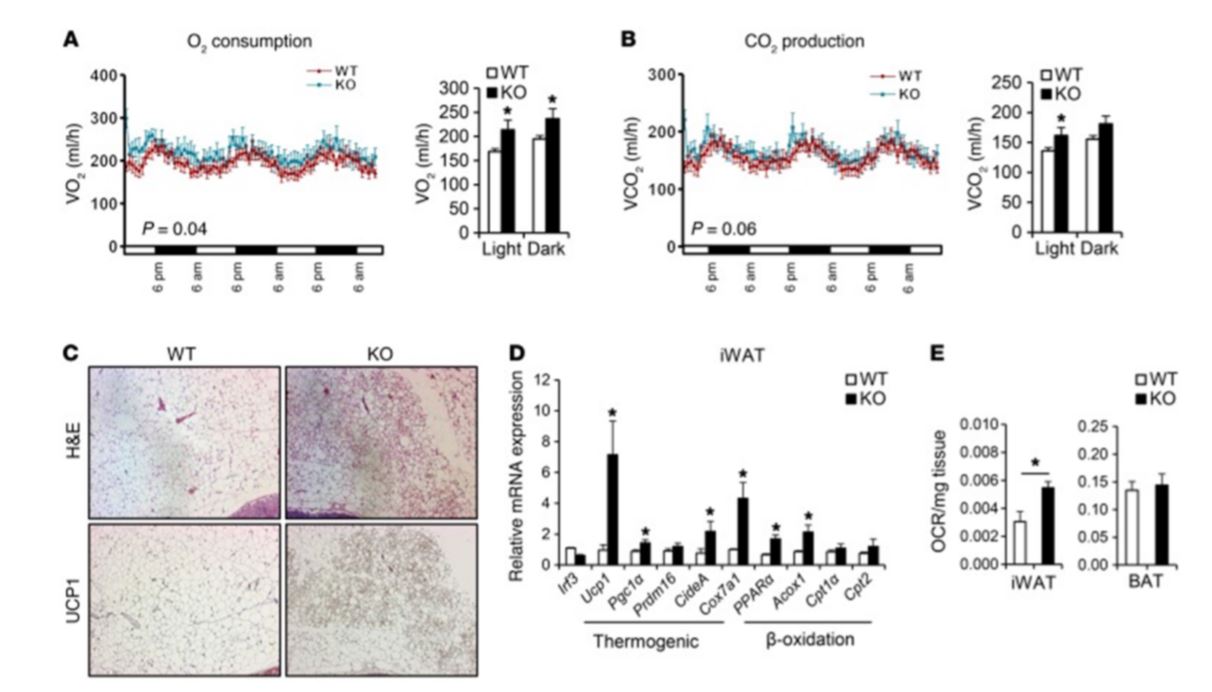
Figure 4 Irf3–/– mice show increased browning of white fat. Male WT and Irf3–/– mice (n = 7–8 mice per group) were placed on HFD for 16 weeks. (A & B) Oxygen Volume (VO2) consumption. (A) Carbon dioxide Volume (VCO2) production (B) Day and night cycle were determined by CLAMS. (C) Histology showing H&E and UCP1 immunostaining in iWAT of WT and Irf3–/– mice on HFD. (D) Expression of thermogenic and β-oxidation genes in iWAT of WT and Irf3–/– mice on HFD. (E) Oxygen consumption rate (OCR) in iWAT and BAT (Brown Adipose Tissue) of WT and Irf3–/– mice on chow diet.
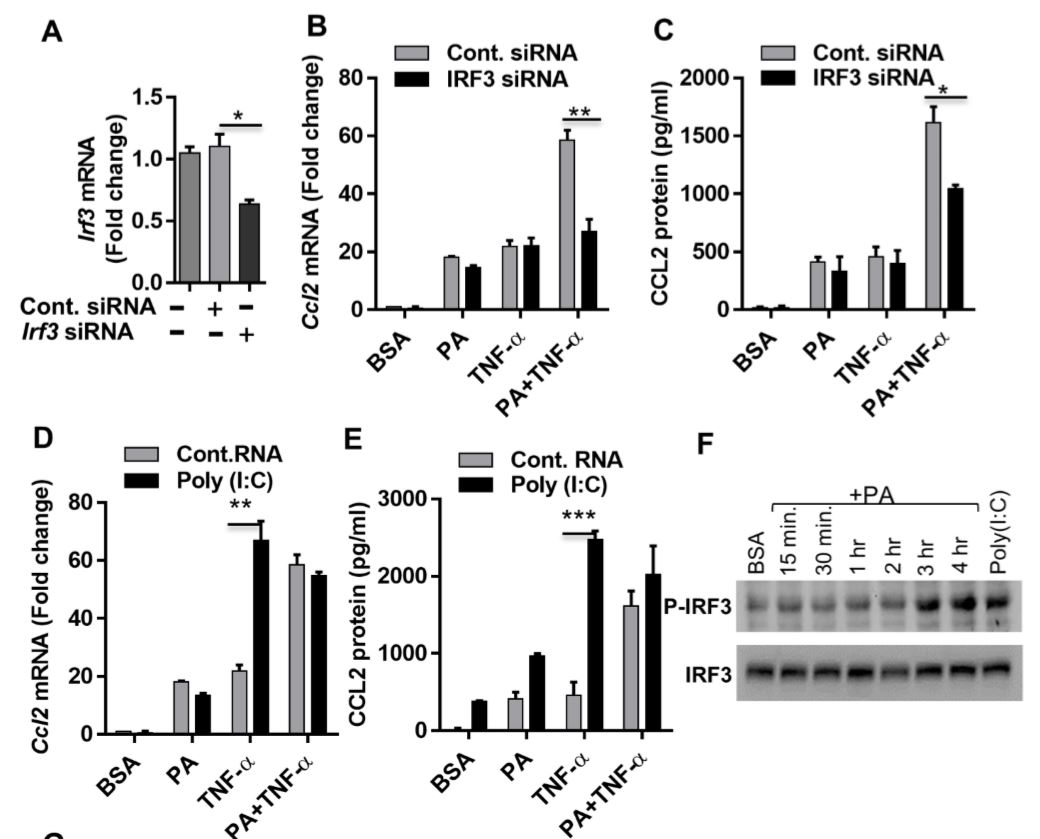
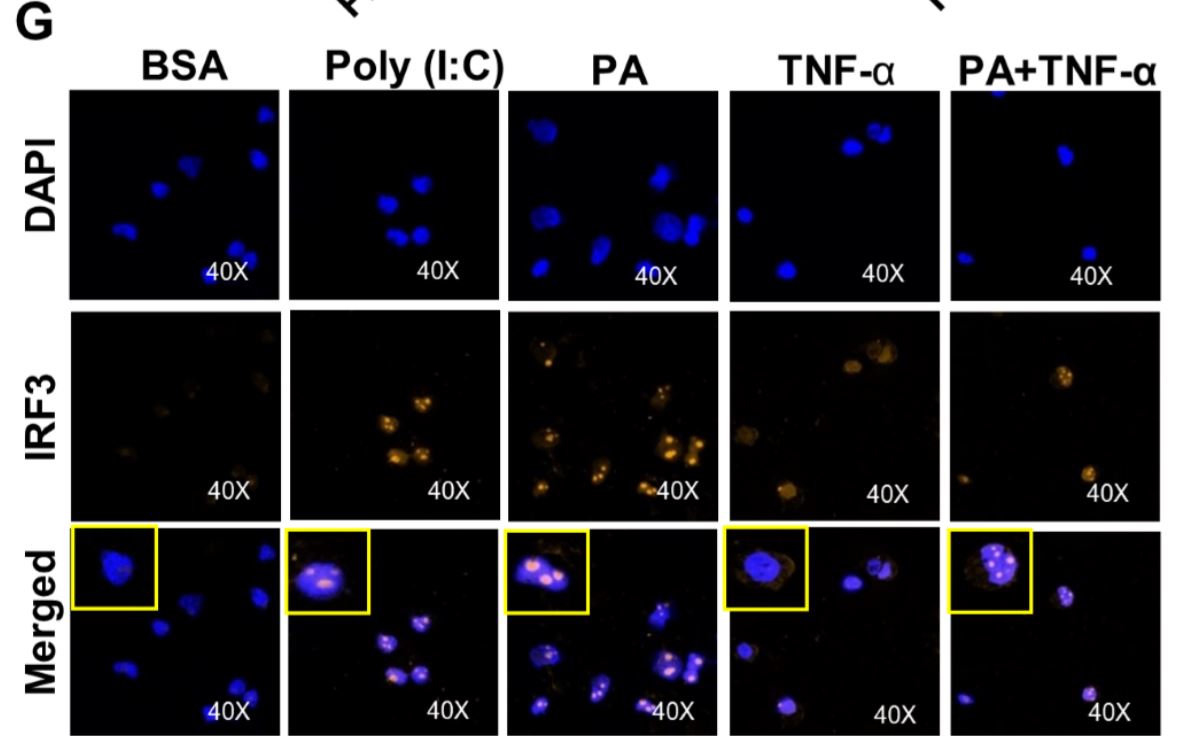
Figure 5 Synergistic production of CCL2 by palmitate (PA)/TNF-α requires IRF3. (A) Monocytic cells were transfected with control or IRF3 siRNA and incubated for 40 h. Real-time PCR was performed to measure IRF3 expression. (B & C) IRF3-deficient cells were treated as indicated, and Ccl2 mRNA and protein expression was determined. (D & E) Poly I:C (an activator of IRF3) increased TNF-α–induced CCL2. Cells were treated (via transfection) with poly I:C for 2 h and then incubated with BSA, PA, or TNF-α, or a combination of PA and TNF-α, for 24 h. (F) Western blot showing phosphorylation of IRF3 after PA or poly I:C treatment. (G) Cells were treated as indicated. Fixed and permeabilized cells were stained for nuclei and IRF3 using DAPI and anti-IRF3 Ab. Confocal laser scanning microscopy showed immunocytochemical localization of nuclei (blue; top panels), IRF3 (red; middle panels), and merged images of nuclei and IRF3 with magnified insets (bottom panels).
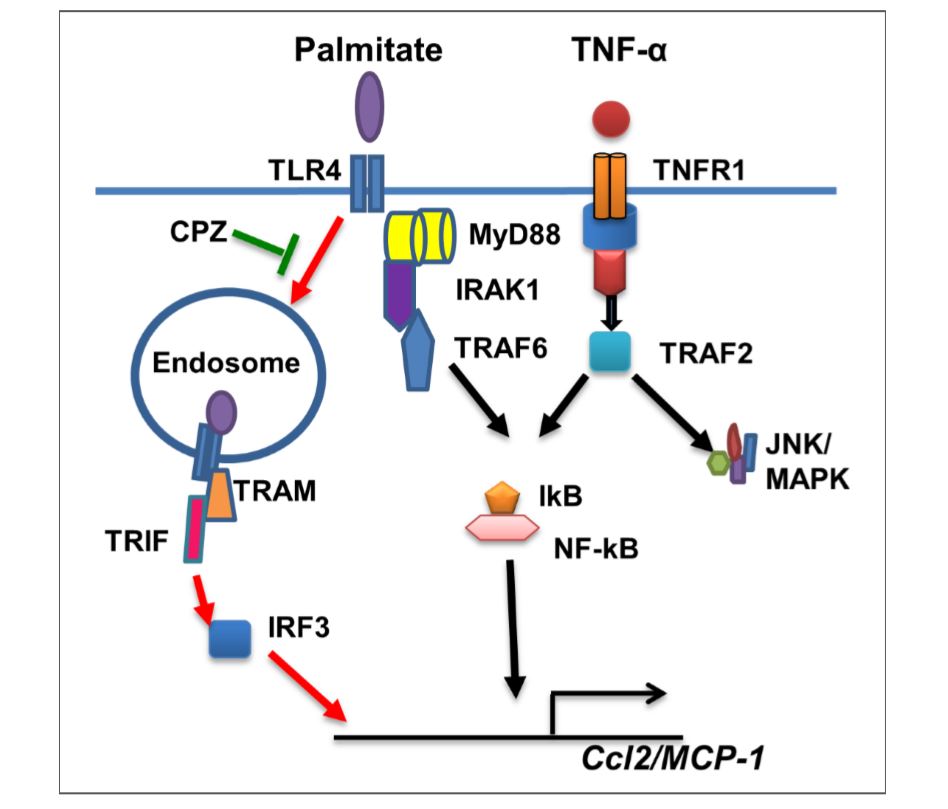
Figure 6 Illustration of signaling pathways underlying the synergy between palmitate and TNF-α for CCL2 production. The pathway highlighted with red arrows represents the predominant mechanism of this synergistic response induced by palmitate. CPZ may inhibit TLR4 internalization and suppress synergy between palmitate/TNF-α for production of CCL2.
